引言
2020年1月15日,中美双方代表在美国华盛顿签署了第一阶段的《中华人民共和国政府和美利坚合众国政府经济贸易协议》(Economic and Trade Agreement Between The Government of The People’s Republic of China and The Government of The United States of America)(简称“《协议》”)。《协议》第一章规定了知识产权的保护和执法方面的问题,其中第1.11条规定了两国在药品专利纠纷早期解决的有效机制(Effective Mechanism for Early Resolution of Patent Disputes)(即药品专利链接制度)方面应作出的改进和完善。首先,作为批准包括生物药在内的药品上市的条件,如果中国允许原始提交安全性与有效性信息的人以外的其他人,依靠之前已经获批产品的安全性和有效性的证据或信息,例如在中国或其他国家、地区已获上市批准的证据,中国应:1、规定制度,以通知专利权人、被许可人或上市许可持有人,上述其他人正在已获批产品或其获批使用方法所适用的专利有效期内寻求上市该产品;2、规定足够的时间和机会,让该专利权人在被指控侵权的产品上市之前寻求第3项中提供的救济;3、规定司法或行政程序和快速救济,例如行为保全措施或与之相当的有效的临时措施,以便及时解决关于获批药品或其获批使用方法所适用的专利的有效性或侵权的纠纷。其次,中国应在全国范围内建立与上述首先部分内容相符的药品相关制度,包括规定专利权人、被许可人或上市许可持有人有权在被指控侵权的产品获得上市许可前提起诉讼,就可适用专利的有效性或侵权的纠纷解决寻求民事司法程序和快速救济。中国还可提供行政程序解决此类纠纷。再次,美国确认,美国现行措施给予与本条款规定内容同等的待遇。
药品专利制度旨在平衡原研药企业与仿制药企业的利益,不仅能够激励药品创新,而且可以促进仿制药发展,从而降低药价,对社会公共健康具有重要意义。本文通过分析中美现阶段药品专利链接制度的立法现状,结合《协议》要求以及我国国情对我国现阶段药品专利链接制度进行评析。
一、 药品专利链接制度概述
药品专利链接制度是指在药品上市审批过程中解决专利纠纷的机制。具体而言,该制度要求药品监管部门在审查药品注册申请批准上市决策时,不仅对“安全性、有效性”进行审查,同时一定程度考察其是否存在侵犯他人专利权的情况。 该制度包含五个基本制度:(一)药品专利登记制度;(二)仿制药专利声明和通知制度;(三)仿制药简略申请制度(ANDA);(四)专利挑战制度(包含专利挑战诉讼和批准等待期);(五)首仿药数据保护期制度。
药品专利链接制度最早起源于美国,其后多个国家相继建立了专利链接制度。我国的药品专利链接制度的建立可追溯到2005年5月1日施行的《药品注册管理办法》,虽然经过一系列的修改和补充,但我国的药品专利链接制度在立法层面依旧处于探索阶段。
二、 美国药品专利链接制度立法现状
美国的专利链接制度始于《Hatch-Waxman法案》,在此之后,通过在实践中不断的发现问题,美国国会对该制度进行了一系列的修改和完善。现阶段美国药品专利链接制度具体而言主要包括以下几方面的内容:
(一) 药品专利登记制度(又称桔皮书制度)
当美国食品和药物管理局(简称“FDA”)批准药品上市后,该药品的所有专利信息以及数据独占保护期将会被记录在桔皮书上,作为仿制药企业提出Abbreviated New Drug Applications(简称“ANDA”)申请的重要依据。
(二) ANDA制度
在ANDA制度下,仿制药企业提出上市申请时,不需要提供证明其安全性和有效性的试验数据(原研药企业提出上市申请时需提供),只需证明仿制药与原研药具有一样的剂型、活性成分、生产规格等信息,从而在一定程度上节省了申请所需的时间和资金。在提交上市申请时,仿制药企业需查阅桔皮书上所记录的原研药的相关信息,并作出声明。根据相关法案规定,声明有以下四种:(1)没有其他相关专利登记在桔皮书上;(2)桔皮书上登记的相关专利已过专利保护期;(3)桔皮书上登记的相关专利即将到期,但仿制药不在其专利保护期内上市销售;(4) 桔皮书上登记的相关专利无效或者提交申请的仿制药不会造成侵权。
当仿制药企业作出第四种声明时,该企业应当在提出申请的20日通知专利权人。专利权人在接到通知的45日内有权向法院提起专利侵权诉讼。超过45日起诉,FDA将会继续审查仿制药企业提起的上市申请。若专利权人在起诉期限内提起专利侵权诉讼,仿制药的上市申请审查将进入“停止期”。在此期间, FDA将会暂停对仿制药的上市申请审查,等待法院判决。“停止期”最长为30个月,若超过30个月未作出判决,法院可以临时批准符合条件的仿制药的继续审查。如果法院判决专利无效或者或者仿制药不会侵犯原研药的专利,那么FDA将会继续进行仿制药的ANDA审查,而后授予首家向FDA提出第四种专利声明的仿制药企业180天的独占期。
为了防止专利权人滥用30个月的“停止期”,美国在后来通过的《更容易获得可支付药品法》里规定了质疑机制,即当专利权人提起专利侵权诉讼后,仿制药企业可以对其提出反诉,主张该药品不应当被列入桔皮书中。若胜诉便可将该原研药从桔皮书中剔除。
(三) Bolar例外制度
Bolar例外制度源于美国联邦巡回上诉法院在1984年对Roche诉Bolar案,后规定在《Hatch-Waxman法案》中。在该制度下,仿制药企业可以在药品专利到期前进口、制造以及使用专利药品进行试验以获取FDA等部门要求的信息,并且该行为不侵犯药品的专利权。
(四) FDA和美国专利商标局(简称“USPTO”)的职能链接
在《Hatch-Waxman法案》中规定,FDA负责审查提出上市申请的仿制药品的安全性和有效性,USPTO负责审查药品是否能够被授予药品专利权(对提出上市申请的药品专利状态进行实质审查)。在这种机制下,药品最终的上市需要经过两个机构的独立审查,两机构通过协调而实现职能链接。
三、 我国药品专利链接制度立法现状及对比
相较于美国,我国对药品的专利保护开始的较晚,药品注册上市与药品专利在以往的很长一段时间是两个完全分割的领域,从而导致了我国药品行业很大一定比例被仿制药占领的情况。2002年通过的《药品注册管理办法(试行)》首次将药品注册与药品专利联系在了一起,在这之后,我国相继出台了一系列的法律法规与政策来加强药品专利保护。其中,药品专利链接制度也逐渐被完善。
(一) 信息公示制度
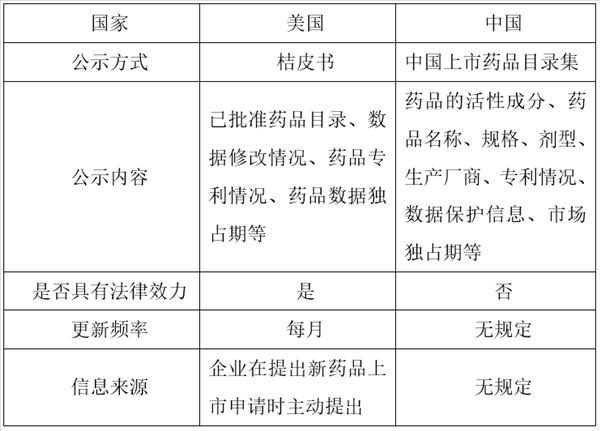
2007年修订通过的《药品注册管理办法》第八条规定了相关部门应当公开已批准的药品目录等信息,但对应当公开的信息内容并未作出具体规定。2017 年国家食品药品监督管理总局(简称“CFDA”) 发布的《中国上市药品目录集》(简称“《目录集》”)正式建立药品登记制度。但该《目录集》与美国的桔皮书相比有一定的不足之处:
(二) 仿制药品注册申请制度
根据《药品注册管理办法》《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》《关于鼓励药品医疗器械创新保护创新者权益的相关政策(征求意见稿)》等相关规定,我国仿制药品注册申请制度与美国的ANDA制度相似。
首先,仿制药企业在提出注册申请时,应当提交专利声明,说明其设计的相关专利及其权属状态,并在规定期间内告知相关药品专利权人。仿制药企业可以通过声明其行为不构成对原研药品专利侵权来挑战原研药专利。挑战成功即可提前上市仿制药,无需等待专利到期。 进行专利挑战的仿制药企业应当在提出申请之日起20日内告知药品专利权人。药品专利权人认为其权利受到侵犯的可在接到申请之日起20日内提出专利侵权诉讼,药品评审机构可设置最长不超过24个月的批准等待期,但在此期间不停止该仿制药的注册审理工作。对于专利挑战成功的仿制药品,我国也给予1.5年的数据保护期(相当于美国该制度下的市场独占期)。
但在该制度下,与美国相关规定不同的主要有两点,其一,在我国,申请人提交专利声明并未要求专利部门介入并对其进行实质性的审查。而在美国,USPTO会对声明中所述专利信息作实质性审查并将审查结果反馈给FDA;其二,在我国,在权利人提出专利侵权诉讼之后,CFDA决定批准最长不超过24个月的“等待期”,且在此期间评审机构并不停止对仿制药品上市申请的审批。
相比美国的“30个月的停止期”来说,此制度设计有一定的优势。首先,在美国,“停止期”的设计过度维护了专利权人的利益。只要权利人提起了专利侵权诉讼,FDA就必须停止审查,这就如同使专利权人获得了临时禁令,进而禁止仿制药在诉讼期间不得上市, 而法院在颁发临时禁令时不仅需要当事人负有一定的举证责任,法院还需对此进行严格的审查。 美国“停止期”的设计使得权利人可不负任何举证责任而取得同样的效果。其次,在美国,只要专利权人提起了诉讼,仿制药的申请就会自动进入“停止期”, 在“ GlaxoSmithKline v Apotex”案中,专利权人最后获得了五次自动“停止期”,导致FDA在第一次ANDA申请日的 65 个月后,才核准第一个仿制药上市。在我国,CFDA具有一定程度的自由裁量权,可自由裁量是否批准“等待期”以及“等待期”的时长(最长不超过24个月)。此规定可以有效的调节“等待期”延缓仿制药上市的遏制效力。
(三) Bolar例外制度
2008年修改通过的《专利法》第69条第5项规定,仿制药企业在原研药专利有效期内,为了审批的需要而制造原研药的,不视为专利侵权,从而原研药的专利到期后仿制药可以立即上市,减少了原研药在实践中的专利垄断期。在最高人民法院2019年判决的石药集团恩必普药业有限公司(简称“恩必普公司”)与丽珠集团利民制药厂(简称“利民制药厂”)侵害发明专利权纠纷案件中,因恩必普公司并未提交证据证明利民制药厂在为提供行政审批所需信息之外,还存在以生产经营为目的另行实施了制造、使用、许诺销售、销售、进口涉案专利产品,或者使用涉案专利方法以及使用、许诺销售、销售、进口依照涉案专利方法直接获得的产品的行为,故认定利民制药厂未侵害恩必普公司的涉案专利权。我国对于这一制度的规定与美国的规定相差不大。
四、 我国药品专利链接制度的待完善之处
《协议》要求中国应建立以下制度:(1)通知专利权或其他利害关系人,其他人正在其专利有效期内寻求上市相关产品;(2)让专利权人有足够的时间和机会在其他人产品上市之前获得司法或行政的快速救济,如行为保全等临时措施;(3)建立专利有效性或侵权纠纷解决的民事司法程序或行政程序和快速救济。我国现有的相关规定还不能全面满足《协议》的要求,我国药品专利链接制度有以下几点需要完善:
(一) 完善信息公示制度
首先,我国的《目录集》并没有法律效力,在司法实践中并不能作为法律依据。虽然国家在各种政策中多次明确指出要建立完备的上市药品目录集,但是缺乏立法层面的支持。
其次,《目录集》中所涵盖的专利信息不全面。目前的《目录集》中仅含有批准文号/注册证号、专利号和专利到期日,并未含有专利权人信息和专利法律状态。 在仿制药品简化申请制度中,仿制药企业需要向原研药企业发出不侵权声明,缺乏专利权人的信息不便于仿制药企业提供声明。同时,缺乏专利法律状态也给仿制药品作出不侵权声明带来了不便,仿制药企业需要根据不同的专利法律状态作出不同的不侵权声明。
再次,《目录集》更新频率不确定。作为药品专利链接制度重要的信息依据,《目录集》所记载的内容需要保持正确性以及及时性;
最后,《目录集》的信息来源不确定。
(二) 完善仿制药品简化申请制度
1. 专利申明审查制度
现阶段,我国CFDA与国家知识产权局(简称“CNIPA”)并未建立职能链接制度。在仿制药品上市时,仅有CFDA进行审查。CFDA仅能对对药品的仿制药与原研药具有一样的剂型、活性成分、生产规格等医药相关信息进行审查,但对申请所涉及到的专利信息并不能做到实质审查,增加了未来专利侵权纠纷产生的可能性。
2. 简化专利无效宣告程序
我国现阶段法院无权宣告专利无效,需要由专利复审委员会作出权利无效的宣告。而该无效宣告是可诉的行政行为,这将会导致审理专利无效类型案件的时间延长,从而使得药品专利链接制度失去其促进药物上市的意义。例如著名的“正大天晴专利挑战案”虽然最后取得胜利,涉案专利被认定为全部无效,但该案历时4年之久,不仅仅是对正大天晴企业财力以及人力的消耗,也在一定程度上降低了涉案药品“恩替卡韦片”在国内的可及性。2001年开始的“吉西他滨专利挑战案”更是耗时9年之久。因此在药品专利链接制度中,应当缩短专利无效宣判时间,更大程度上发挥药品专利链接制度促进药物上市的作用。
(三) 适当结合国情
一套完备的制度在实际运行过程中是否能够达到预期的效果一定程度上取决于制度的制定是否符合一国具体国情。药品专利链接制度的设计不仅要参考“制度先行者”的设计,还需要结合我国国情。具体而言,需要考虑以下几个方面:其一,美国作为药品研发大国,原研药在药品市场占据很大份额。相反,我国现阶段药品市场中仿制药占比较高。在制度设计中要充分考虑这一因素,避免过多保护原研药企业利益而造成对药品市场的过度冲击。其二,与美国不同,我国实行的是全面医保制度,药品的价格在一定程度上受国家调控,不会造成原研药价格过高。
小结
《协议》对专利纠纷早期解决机制提出了更高的要求,我国现阶段的法律制度建设尚未完全匹配,还需进一步对该制度进行补充和完善。美国在药品专利链接制度方面走在了我们的前面,我们需结合我国药品行业实际情况、法律体系以及社会福利体系等,综合构建既符合各方利益平衡需求,又符合我国国情的药品专利链接制度。尤其在新型冠状病毒疫情期间,这个问题的重要性愈发凸显。
邵阳专利知识律师事务所(www.tieqiaolawyer.com/zhuanlizhishi)提供邵阳市专利知识24小时在线免费咨询
J&T资本观察 | 注册制2.0时代的IPO股东核查要求
2025/4/15 6顾金龙:面对股权回购,创始人应当知道的谈判要点
2025/4/13 79不满补偿金额律师助力起诉,镇政府已多次提出协商沟通
2025/4/13 71春节期间法定加班计算加班费的依据是什么
2025/4/13 75新《北京市物业管理条例》亮点——业主篇(下)
2025/4/13 72关于当事人协商解决后如何追究侵权人行政法律责任的批复
2025/4/13 69最高法院:关于婚姻案件诉讼程序、共同财产等48个实务问答(收藏)
2025/4/13 782023劳动合同范本免费,劳动合同书样本
2025/4/13 72国内养老信托的主要法律模式解析
2025/4/13 80最高院、最高人民检察院关于办理利用互联网等复制...
2025/4/13 76信•条——非法采矿罪与非罪的重构
2025/4/13 70我国首部个人破产法规在深圳施行!首家个人破产事务
2025/4/13 74【新政解读】云南新一轮征收开始!2023年玉溪政府批复,玉溪市这些地方要征地
2025/4/13 74门店合同到期租客不搬怎么办?
2025/4/13 68